Introducing dual assembly
Definition. The dual assembly of a diploid sample consists of two sets of contigs with each set representing one complete haploid genome. Similar to contigs in a primary assembly, contigs in a dual assembly may have occasional switches between parental haplotypes. I called such an assembly as partially phased assembly in an earlier post but decided to coin a new term in our new hifiasm preprint for clarity.
Why dual assembly?
A primary or collapsed assembly only represents one haploid genome. It is okay and may be preferred if we want to construct the reference genome of a new species. However, if we want to profile sequence variations in a population, such an assembly would not work as it randomly misses half of information in a diploid genome. It is necessary to recover both haplotypes.
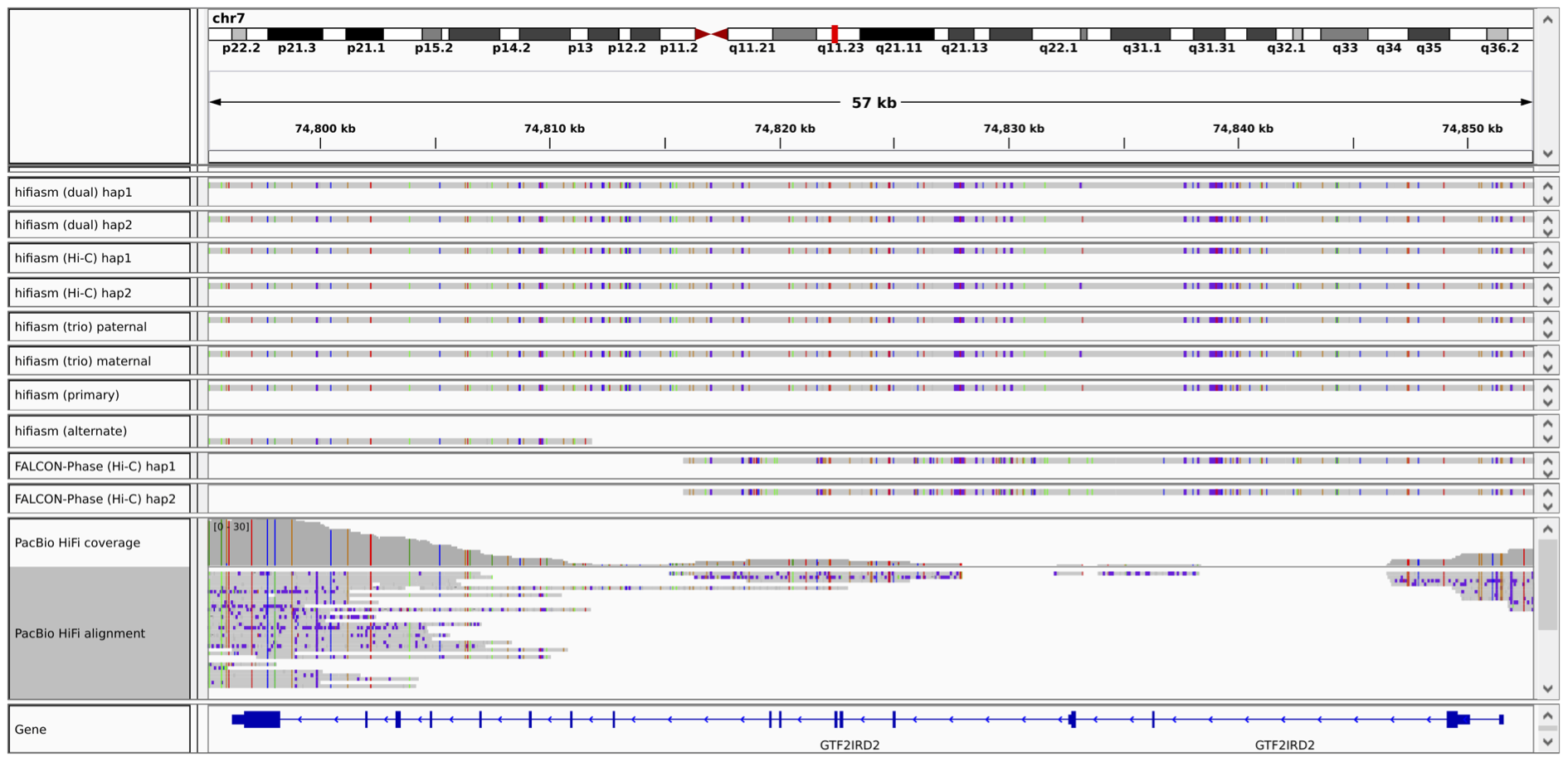
Haplotype-resolved de novo assembly is the ultimate solution to variant calling. Genome-In-A-Bottle (GIAB) recently constructed two new variant calling benchmarks, one on HLA and the other on clinically important genes. In both cases, GIAB used haplotype-resolved assembly as the main source of ground truth because assembly can reconstruct complex regions longer than the read length. In this case, reference-based read mapping would often misplace reads due to the lack of long-range information. The figure below shows an example where read mapping leaves a gap in gene GTF2IRD2 while trio or Hi-C assemblies can go through both haplotypes.
However, producing a haplotype-resolved assembly requires multiple data types, such as parental sequences or Hi-C in addition to long sequence reads. This increases sequencing costs and is sometimes infeasible, for example, when we cannot obtain enough DNA or don’t have access to parental samples. The need of more data is particularly problematic for clinical samples.
A dual asembly can be produced with long reads only. It is a weaker version of haplotype-resolved assembly but is almost as powerful for variant calling purposes. In the figure below, the dual assembly (the top two tracks) also correctly resolves both haplotypes. The primary/alternate assembly pair can also be produced with long reads only. However, the alternate assembly is often too fragmented. It misses one haplotype in the example below.
I recommend to produce a dual assembly for the calling of structural variations, if you only have long reads. As of now, only hifiasm and peregrine can do such assembly for PacBio HiFi data only but I expect more assemlers to support such assembly for more data types.
blog comments powered by Disqus