Evaluating collapsed misassembly with asmgene
Why?
It is usually easy to evaluate the contiguity of a de novo assembly – just compute N50. It is much harder to evaluate the correctness. We typically identify misassemblies by aligning contigs to a reference genome. However, it is tricky to interpret the results. In case of human, there are thousands of structural variations (SVs) between the reference and the sample being assembled. Alignment-based evaluation often mistakes these SVs as misassemblies. For example, QUAST identifies >10,000 “misassemblies” in the T2T assembly when compared to GRCh38. We can’t reliably tell misassemblies from SVs which leads to overestimated misassembly rate. A second problem with reference-based alignment is that most alignment differences come from complex regions such as centromeres and subtelomeres. It fails to evaluate gene regions we are mostly interested in; on the contrary it penalizes an assembly that represents these complex regions better.
How?
Most assembly problems are caused by repetitive or paralogous regions. When an assembler cannot resolve such a region, it either creates an assembly gap or forces through the region with a misassembly. To probe these issues, we can align a multi-copy gene to the assembly and see if it remains multi-copy.
More precisely, we do the following. We align all annotated transcripts to a reference genome and select the longest isoform among overlapping transcripts. For each selected transcript, we record a hit if the transcript is mapped at ≥99% identity over ≥99% of of the transcript length. A transcript is considered to be single-copy (SC) if it has only one hit; otherwise it is considered multi-copy (MC). We do the same for the assembly and then compute the fraction of missing multi-copy gene as
MMC = 1 - |{MCinASM} ∩ {MCinREF}| / |{MCinREF}|
In the ideal case of a perfect
assembly, %MMC should be zero. A higher fraction suggests more
collapsed assemblies. We can compute percent MMC (%MMC) with paftools.js
asmgene from minimap2:
minimap2 -cxsplice:hq -t8 ref.fa cdna.fa > ref.cdna.paf
minimap2 -cxsplice:hq -t8 asm.fa cdna.fa > asm.cdna.paf
paftools.js asmgene [-a] ref.cdna.paf asm.cdna.paf
The output looks like:
H Metric ref.cdna asm.cdna
X full_sgl 36426 36389
X full_dup 0 18
X frag 0 4
X part50+ 0 5
X part10+ 0 0
X part10- 0 10
X dup_cnt 1334 1330
X dup_sum 4110 4080
On the line X dup_cnt, 1334 is the number of multi-copy genes in the
reference, of which 1330 remain multi-copy in the assembly. %MMC is thus
1-1330/1334=0.3%. Also in this output, 36426 is the number of single-copy genes
in the reference, of which 36389 remain single-copy in the assembly and 18 are
false duplications. We can similarly compute BUSCO-like metrics but
based on the reference.
Collapsed misassemblies in long-read assemblies
The following figure shows the level of collapsed genes in various CHM13 assemblies, taking Ensembl genes as input and the T2T CHM13 as the reference:
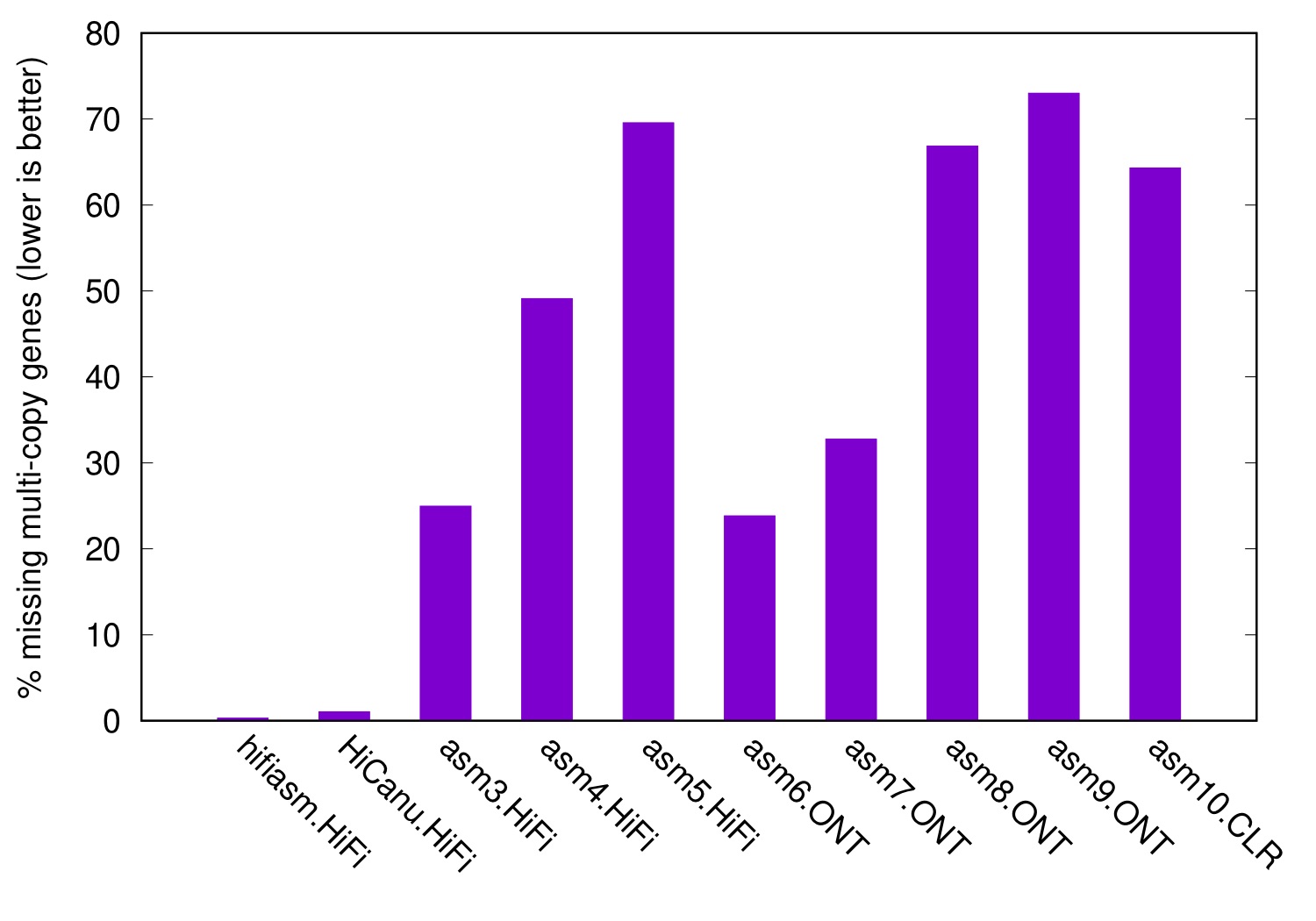
Because the reference and the assemblies all come from the same sample, a perfect assembly should have no collapsed genes. Hifiasm and HiCanu are close to that mark. However, some assemblers may miss up to 70% of multi-copy genes. We had a closer look at missing genes. As is expected, they either fall into assembly gaps or leave a misassembly in a long contig. If you want to study a gene family, such assembly problems will ruin your day.
CHM13 is a homozygous cell line. The following figure shows the level of collapsed genes in diploid HG00733 assemblies, again with CHM13 as the reference:
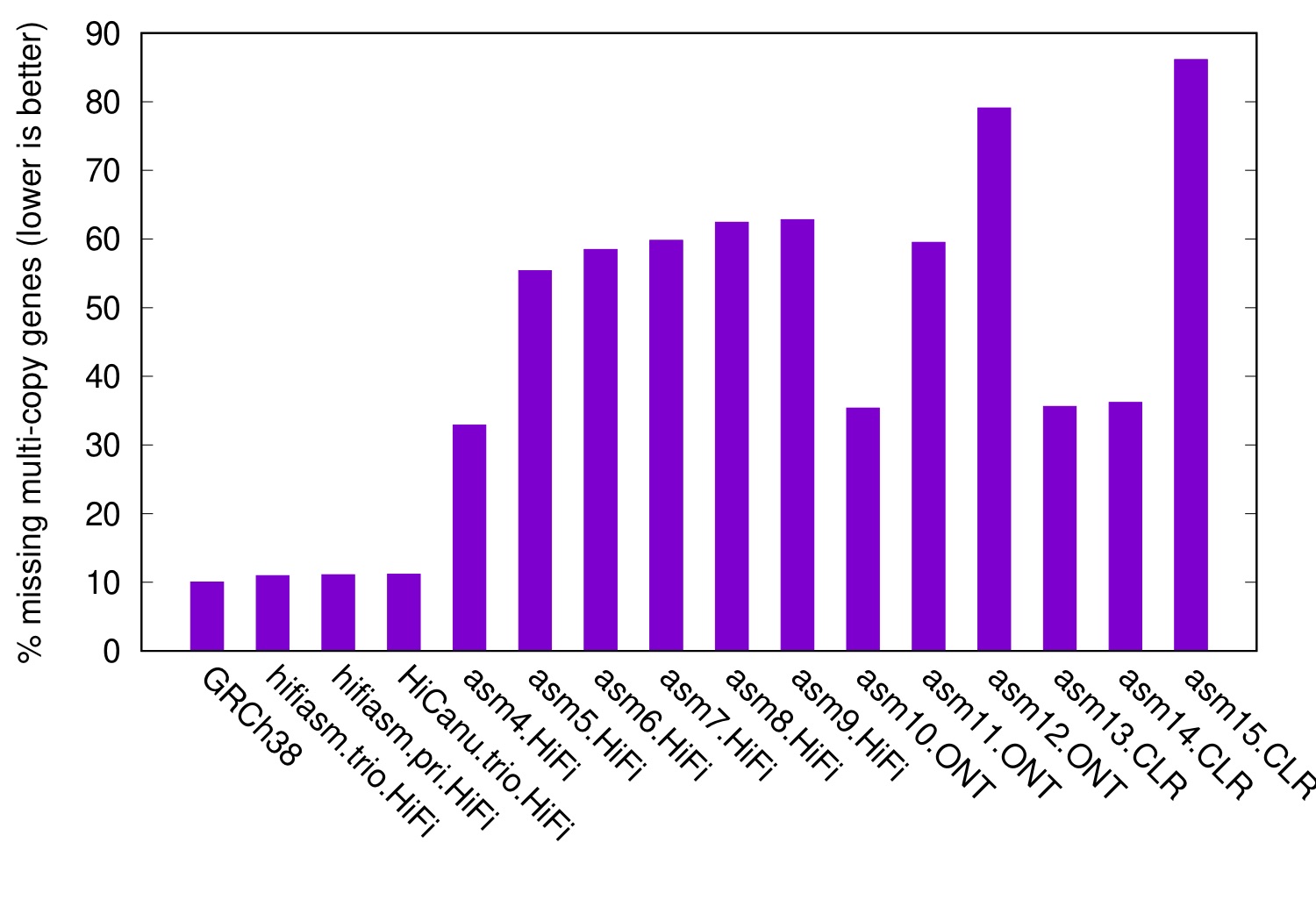
In this figure, even GRCh38 misses 10% multi-copy genes in CHM13. This is background noises caused by between-sample SVs. It is much lower than the level of collapsed misassemblies of many assemblers, demonstrating the effectiveness this metric.
Conclusion
Percent MMC is a new metric to measure the quality of an assembly. It takes minutes to compute, is gene focused and is robust to structural variations in comparison to evaluations based on assembly-to-reference alignment. The obvious downside of %MMC is that it requires a high-quality reference genome and is not applicable to new species, but this is not a concern during the development of assemblers.
blog comments powered by Disqus